GENERALIDADES Y TRATAMIENTOS EMERGENTES EN LA
BETA-TALASEMIA
OVERVIEW AND NEW TREATMENTS IN BETA-THALASSEMIA
Guido Jose Angulo 1 Rebecca Koss Hernández 2 José Manuel Monge Ortiz 3
1.Médico general Hospital CIMA San José Costa Rica
2 y 3 Médico General trabajador independiente, San José Costa Rica
Contactos: guidono123@gmail.com rebecakoss@gmail.com mongeortiz@gmail.com
RESUMEN
La beta-talasemia es un trastorno congénito causado generalmente por mutaciones
puntuales en el gen que codifica para la síntesis de cadenas beta de la globina, lo
que produce un fenómeno de eritropoyesis ineficaz y reducción de la vida media
de los eritrocitos en las formas más severas. Dentro de la clasificación se engloban
formas de talasemia de diversos comportamientos clínicos, desde formas severas
hasta formas asintomáticas. Las complicaciones de las talasemias muchas veces se
asocian al tratamiento crónico de la misma. El tratamiento actual conlleva transfu-
siones sanguíneas a repetición, entre otros. El desarrollo de nuevas terapias dirigi-
das mitiga los efectos adversos asociados a las transfusiones, además de intervenir
directamente en la parte genética.
Palabras clave: talasemia; hemoglobinas; terapia genética; transfusión sanguínea;
quelantes del hierro.
ABSTRACT
Beta-thalassemia is a congenital disorder generally caused by point mutations in
Cómo citar:
the gene that encodes for the synthesis of globin beta chains, which produces an
ineffective erythropoiesis phenomenon, and reduces the half-life of erythrocytes in
Angulo, G., Koss Her-
the most severe forms. However, the classification includes forms of thalassemia of
nández, R., & Monge
Ortiz, J. (2021). Ge-
various clinical behaviors; from severe forms to asymptomatic forms. Complications
neralidades y trata-
are usually associated with the chronic treatment utilized in this disorder, which in-
mientos emergentes
cludes repeated blood transfusions, amongst others. New therapies seek to reduce
en la Beta-Talasemia. the adverse effects associated with transfusions, as well as targeting genetic com-
Revista Ciencia Y Sa-
ponents of the disease.
lud, 5(1), Pág. 47-58.
Keywords: thalassemia; hemoglobins; genetic therapy; blood transfusion; iron che-
lating agents.
18/nov/2020
15/ene/2021
4
47
INTRODUCCIÓN
Las beta-talasemias son un grupo de anemias hereditarias heterogéneas, de herencia autosómica recesiva,
caracterizadas por una síntesis de cadena beta-globina reducida o ausente (1). Este grupo de enfermedades
presenta una clínica y severidad variable, siendo los pacientes con beta-talasemia mayor, los más afectados.
En su condición más severa los sujetos presentan anemia y expansión de la médula ósea. Sin tratamiento, la
beta-talasemia conduce a hepatoesplenomegalia, deformidades óseas debido a la expansión de la médula
ósea extra medular e insuficiencia cardíaca debido a anemia severa (2).
Desde la primera descripción de las talasemias severas, publicada hace más de 90 años por Cooley y Lee,
los avances en el entendimiento de esta enfermedad han sido notables. Dada su prevalencia relativamente
elevada, los estudios en la detección y tratamiento de las beta-talasemias han arrojado nuevos entendi-
mientos sobre génesis.
La extensa comprensión de la fisiopatología de estas enfermedades ha permitido un aumento en la super-
vivencia y calidad de vida de los pacientes con beta-talasemia en las últimas décadas (3).
El objetivo del presente artículo es exponer de manera concreta la patogénesis y clínica de la beta-talase-
mia, para hacer énfasis en terapias emergentes y adyuvantes al tratamiento actual existente y su posible
utilidad en el futuro.
MÉTODOS
Se realizó una revisión exhaustiva de bases de datos de ScienceDirect, Elsevier, PubMed y Access Medicine.
Dado que se quieren exponer aquellas terapias emergentes y más novedosas, se restringió la antigüedad
de los artículos consultados a no más de cinco años, cuyo énfasis fuese además tratamiento y desarrollo
de nuevas terapias. Asimismo, se utilizaron libros de textos especializados en hematología para establecer
conceptos básicos.
Se encontraron más de 70 artículos relacionados con el tema, de los cuales se eligieron 15 debido a su en-
foque en terapias emergentes y tratamientos a futuro. Así mismo, se consultaron dos textos especializados
en hematología en sus más recientes ediciones.
1.
EPIDEMIOLOGIA
Junto con la drepanocitosis, las talasemias son las enfermedades monogénicas más comunes a nivel mun-
dial (4). Se estima que de 80 a 90 millones de personas son portadoras de esta enfermedad en todo el
mundo (1,5% de la población mundial) (1).
Aunque la epidemiología de las diversas formas clínicas sigue siendo poco conocida, se sabe que la enfer-
medad es altamente prevalente en el área que se extiende desde África subsahariana, a través de la región
mediterránea y Medio Oriente, hacia el subcontinente indio y el sudeste asiático. De esta manera, más del
90% de los pacientes con estos trastornos viven en países de bajos y medianos ingresos (3).
2.
ETIOLOGÍA
Los síndromes talasémicos corresponden a un grupo de anemias hemolíticas de tipo hereditarias, causadas
por defectos congénitos en la biosíntesis de las cadenas de globina; en esta revisión en particular, se trata
de la cadena beta de la hemoglobina (5).
48
La beta-talasemia se puede subclasificar en tres grupos de acuerdo con el comportamiento clínico y mu-
taciones presentes:
1.
Beta-talasemia mayor o Anemia de Cooley
2.
Beta-talasemia intermedia
3.
Beta-talasemia menor, portador o rasgo talasémicos beta
3. PATOGÉNESIS
La hemoglobina corresponde a la principal molécula transportadora de oxígeno en sangre compuesta por
un tetrámero de dos cadenas de polipéptidos. La hemoglobina adulta (HbA) se compone de dos cadenas
alfa y dos cadenas beta. En la beta talasemia sucede una menor síntesis de cadena beta de globina, con
la consecuencia de una menor producción de tetrámeros de hemoglobina normales. Las mutaciones en
los genes productores de cadenas beta, en lo que respecta a beta-talasemias, se pueden clasificar en dos
grupos:
-
Mutaciones Beta0: eliminan en su totalidad la producción de cadenas beta.
-
Mutaciones Beta+: eliminan de manera parcial o incompleta la producción de cadenas beta.
En la beta-talasemia mayor, la producción de las cadenas beta es ausente en su totalidad. Por lo tanto, no
se puede formar el tetrámero normal de hemoglobina y la cantidad de HbA se encuentra profundamente
disminuida. Estos pacientes pueden ser homocigotas para la mutación beta0, o poseer una mutación beta0
y una beta+.
En la beta-talasemia intermedia, la producción de cadenas beta se encuentra disminuida pero no comple-
tamente ausente. Estos pacientes presentan dos mutaciones beta+. Los síntomas presentes suelen variar
de acuerdo con la mutación presente.
La beta-talasemia menor o rasgos talasémicos presentan un gen normal y una mutación beta+. Esto suele
ser un hallazgo incidental, aunque puede ser sintomático en algunos pacientes (5).
El aspecto que más repercusiones tiene clínicamente en cuanto a morbimortalidad en estos pacientes es
la acumulación de subunidades de cadenas de globina, en este caso, de la cadena alfa. Estas cadenas son
inestables en la ausencia de las cadenas beta. Por ende, forman precipitados que propician la generación
de radicales reactivos de oxígeno y daño a la membrana de los eritrocitos. Esta anomalía produce hemólisis
y alteración en la maduración celular (6).
Debido a la eritropoyesis ineficaz y sobre todo en pacientes no transfundidos apropiadamente, la anemia y
la hipoxia regulan a la baja la principal molécula reguladora del balance de hierro -la hepcidina-, de forma
que aumenta de forma excesiva la absorción intestinal de hierro, a la vez que ocurre sobrecarga de hierro
sistémica (1).
4. MANIFESTACIONES CLÍNICAS
Las repercusiones clínicas o fenotipos son variables, y los pacientes pueden experimentar desde una ane-
mia crónica severa y dependiente de transfusiones, hasta un estado asintomático (en el caso del estado
portador). Los síntomas se deben principalmente a tres componentes: eritropoyesis ineficaz, anemia cróni-
ca y sobrecarga de hierro (7).
El switch en la síntesis de cadena gamma a cadena beta ocurre desde la vida intrauterina, pero es hasta los
4 - 6 meses de vida extrauterina que el infante depende de la funcionalidad de la hemoglobina del adulto
(HbA) (5).
49
Por ende, los pacientes con beta talasemia mayor usualmente ameritan las primeras intervenciones médicas
desde los 6 meses de edad aproximadamente, con requerimiento de transfusiones de glóbulos rojos para
su supervivencia (1).
Los primeros signos clínicos en pacientes con beta-talasemia mayor incluyen falla para progresar, diarrea,
irritabilidad, infecciones a repetición y esplenomegalia. Sin embargo, sin el abordaje terapéutico apropiado,
las complicaciones de la enfermedad pueden llevar al niño a experimentar: retraso en el crecimiento, pali-
dez, ictericia, visceromegalias, úlceras en miembros inferiores y anormalidades esqueléticas por aumento
en la eritropoyesis extramedular. Usualmente, estos pacientes presentan una anemia profunda (menor a 3
o 4 g/dL) y son dependientes de transfusión (1,5).
En la beta-talasemia intermedia, los síntomas pueden variar en función de la mutación que se encuentre
presente. Estos pueden ir desde anemias leves, trastornos endocrinos (diabetes mellitus) y osteoporosis
hasta tumores extramedulares hematopoyéticos, trombosis y carcinomas hepatocelulares, así como ane-
mias profundas como las observadas en beta-talasemia mayor. La diferencia con esta última radica en que
usualmente estos pacientes suelen no ser dependientes de transfusión desde tempranas edades y en mu-
chos casos los síntomas se desarrollan entre la tercera y cuarta década de vida (8).
En la beta talasemia menor, los pacientes suelen ser asintomáticos. El signo más común es la anemia, que
usualmente es un hallazgo incidental (9).
5. DIAGNÓSTICO
El diagnóstico inicia desde la sospecha clínica. Los síntomas y signos clínicos en las formas de beta talase-
mia mayor y beta-talasemia intermedia con síntomas importantes (anemia con Hb menor a 3-4 g/dL y sus
síntomas asociados) inician antes de los dos años. De esta forma, se debe sospechar esta condición en pa-
cientes en dicho grupo etario con anemia microcítica e hipocrómica severa, visceromegalias e ictericia (1).
El hemograma típicamente muestra anemia severa en rangos menores a 7 g/dL, y un frotis con eritrocitos
microcíticos, poiquilocitos, y eritroblastos. (CAO); estos últimos en relación con el grado de anemia. Tam-
bién se puede realizar análisis por electroforesis o cromatografía para detallar la distribución de hemoglo-
binas en el paciente.
FIGURA 1. Distribución de Hemoglobinas en la electroforesis (1). Gráfico de elaboración por José Manuel Monge Ortiz, autor del ar-
tículo, 2020. NOTA: Las siglas B+ corresponden a beta talasemia con ausencia parcial de cadenas beta y B0 ausencia completa de
cadenas beta.
50
Como se observa en la figura 1, en las formas más severas hay ausencia completa de hemoglobina del adul-
to pues no hay síntesis de cadenas beta y aumento compensatorio en la síntesis de hemoglobina fetal y A2
que no requieren de cadenas beta.
Entonces, ante estos hallazgos en la práctica clínica con niveles aumentados de HbA2 (mayor a 3,5%) se
sospecha un trastorno dentro del espectro de beta talasemias (9).
También se puede hacer diagnóstico molecular basado en las mutaciones que ocurren más frecuentemen-
te. En la mayoría de los casos, las beta talasemias ocurren por mutaciones puntuales (5).
Se puede recurrir a técnicas de reacción en polimerasa de cadena o secuenciación del gen para determinar
las mutaciones más frecuentes. Sin embargo, el diagnóstico en la práctica clínica habitualmente se realiza
con base en las características clínicas, hemograma y electroforesis de hemoglobina (1).
5. MANEJO ACTUAL Y CARGA DE LA ENFERMEDAD
Los pacientes con beta-talasemia mayor o beta talasemia intermedia con síntomas graves requieren tera-
pia de por vida para prevenir y manejar las consecuencias clínicas de la enfermedad; la adherencia estricta
al tratamiento es esencial. Uno de los pilares actuales es la terapia con transfusiones, la cual proporciona
eritrocitos normales y suprime la eritropoyesis ineficaz. Los pacientes con afectación severa requieren
transfusiones de sangre regulares de por vida administradas cada 2 a 5 semanas para mantener los niveles
de hemoglobina entre al menos 9,0-10,5 g/dL. El inicio de esta terapia previo a los 2 años de edad permite
un crecimiento adecuado y evita la deformación ósea asociada a la expansión medular en la mayoría de
pacientes (10).
Actualmente, aproximadamente 100,000 pacientes reciben transfusiones regulares para la beta-talasemia
en todo el mundo. Esto resulta en una carga económica y social no solo para el país, sino además un riesgo
adicional para el paciente. La sobrecarga de hierro de las transfusiones a largo plazo es una causa importan-
te de morbilidad y es la principal causa de mortalidad en pacientes con beta talasemia mayor. Los depósitos
de hierro se dan en el hígado (fibrosis hepática), el corazón (cardiomiopatías e insuficiencia cardíaca) y el
sistema endocrino (hipogonadismo, retraso del crecimiento, infertilidad, diabetes, hipotiroidismo). También
llevan a la disfunción orgánica de los mismos (2)(11).
Las transfusiones además exponen al paciente a riesgos tales como infecciones (si bien los métodos de
cribado actuales son altamente efectivos, el riesgo persiste) y aloinmunización (ocurre en el 10 al 20% de
los pacientes con talasemia).
Debido a las múltiples complicaciones de la sobrecarga férrica, el tratamiento con quelantes de hierro se ha
vuelto otro pilar importante del manejo en estos pacientes. El uso a largo plazo de estos quelantes mejora
las concentraciones de hierro en el hígado y el miocardio; y puede mejorar la función endocrina. Actual-
mente hay tres de estos fármacos aprobados para el uso en beta talasemia: deferoxamina, deferiprone y
deferasirox. El éxito de estos medicamentos no ha sido completo, ya que todavía se presentan problemas
de farmacocinética y de seguridad que muchas veces implican un monitoreo estricto (10).
La esplenectomía es un tratamiento usado clásicamente de manera adjunta a las transfusiones y algunos
estudios han demostrado mejoras en el crecimiento, la calidad de vida y concentración de hemoglobina
para algunos pacientes. Sin embargo, hay cada vez más evidencia de los efectos adversos de este procedi-
miento (riesgo de trombosis venosa, hipertensión pulmonar e infecciones severas), por lo que actualmente
se usa cada vez menos (1)(3).
Actualmente, el trasplante de células madre hematopoyéticas ofrece un enfoque terapéutico potencial-
mente curativo para pacientes con beta-talasemia. Casi el 90% de los pacientes que se someten a este pro-
cedimiento en centros experimentados europeos, sobreviven. Además, la calidad de vida mejora de manera
51
significativa. Mejoras en el manejo de la enfermedad del injerto versus huésped han permitido el uso de
donantes no relacionados y de sangre de cordón umbilical como fuente de células madre hematopoyéticas
para pacientes que no tienen un donante compatible (3)(10).
A pesar de ser una gran alternativa en muchos pacientes, el trasplante de células madre hematopoyéticas
todavía conlleva ciertos riesgos. El uso de regímenes mieloablativos intensos que no siempre evitan el
rechazo del trasplante es una de las principales preocupaciones actuales (9). La mortalidad de este proce-
dimiento se registra entre el 5 y el 10%. Además, es importante recalcar el elevado costo económico que
implica tanto el procedimiento como el seguimiento de los pacientes (3).
6. TRATAMIENTOS EMERGENTES
6.1 INDUCCIÓN FARMACOLÓGICA DE LA HEMOGLOBINA FETAL
Pequeñas cantidades de Hb fetal (HbF) persisten en la edad adulta; sin embargo, representan menos del
1% de la Hb total en la mayoría de los adultos. Los niveles de HbF pueden exceder este umbral en algunos
pacientes con beta talasemia. Se ha observado que estos individuos con altos niveles de producción de HbF
tienen un curso clínico más leve que otros pacientes con esta enfermedad y muchos no requieren transfu-
siones. Es por esto que una importante rama del manejo se ha enfocado en reactivar la expresión génica del
gen de la gamma globulina (abordado posteriormente) y en incrementar la producción de HbF (7).
6.1.1 HIDROXIUREA
La hidroxiurea (HU) es un inhibidor del ciclo celular de la fase S y ha sido probado como clínicamente efec-
tivo en pacientes con anemia de células falciforme. También es beneficioso para algunos pacientes con
beta-talasemias intermedias y reduce la necesidad de transfusiones en un subconjunto de individuos con
beta-talasemias mayor (7).
La terapia con HU lleva a un aumento de 2 a 9 veces en la expresión del ARN mensajero de la cadena gam-
ma, en pacientes con talasemia. Sin embargo, aumentos en los niveles de HbF no siempre se correlaciona-
ron con aumentos en la Hb total en estudios clínicos.
En estudios más pequeños, la terapia de HU también se asoció con mejoras en la función endocrina, úlceras
en las piernas y hematopoyesis extramedular (12).
A pesar de los resultados favorables, este tratamiento se ha asociado a ciertos efectos adversos, incluyendo
citopenias, hiperpigmentación, aumento de peso, infecciones oportunistas, azoospermia en aproximada-
mente el 80% de los hombres (incluso años después de la suspensión del tratamiento) e hipomagnesemia.
Por esto que el uso de HU todavía está restringido a pequeños subgrupos de pacientes (talasemia inter-
media, talasemia Hb-E o pacientes con esplenectomía) y se requieren estudios más a largo plazo sobre su
eficacia (7).
6.1.2 METFORMINA
La metformina es un medicamento aprobado por la Administración de Drogas y Alimentos de los EE.UU.
para el tratamiento de la diabetes mellitus tipo 2. Estudios preclínicos recientes han demostrado que la
metformina induce la expresión de hemoglobina fetal en cultivos de células eritroides primarios y además
tiene un efecto aditivo con la hidroxiurea. Actualmente, hay un estudio en fase I con metformina en la enfer-
medad de células falciformes y en la beta-talasemia no dependiente de transfusiones (NCT02981329) que
incluye niños de 12 años y mayores (6).
52
6.1.3 TALIDOMIDA/LENALIDOMIDA
Los estudios in vitro han demostrado que estos compuestos disminuyen la maduración eritroide, aumentan
la proliferación de eritroides inmaduros y regulan la transcripción de genes de globina, lo que resulta en
inducción de HbF. Algunos estudios iniciales de la talidomida han demostrado beneficios aumentando la
producción de HbF en pacientes con talasemia, aunque el riesgo-beneficio aún necesita establecerse en
vista de la toxicidad potencial de estos medicamentos (12).
6.1.4 SIROLIMUS
El sirolimus es otro fármaco con potencial de inducción para la producción de HbF en pacientes con talase-
mia, ya que también aumenta la expresión de RNAm de la cadena gamma, por lo tanto, lleva a un aumento
correspondiente en HbF. El riesgo-beneficio de este medicamento debe ser confirmado en grandes estu-
dios de pacientes (12).
6.1.5 OTROS
Además de los fármacos mencionados previamente, se encuentran bajo investigación drogas como 5-aza-
citidina, decitabina (hipometilantes) y derivados de ácidos grasos de cadena corta, los cuales han tenido
resultados alentadores en ensayos clínicos. Estos agentes inducen HbF por diferentes mecanismos que aún
no están bien definidos. Su potencial en el manejo de la beta-talasemia está bajo investigación todavía (13).
6.2 AGENTES MODULADORES DE LA ERITROPOYESIS
Las moléculas GDF11 y GDF15 son parte de la familia de moléculas de crecimiento transformante beta y son
parte de la fisiopatología de la eritropoyesis ineficaz (6), aunque con un mecanismo poco claro todavía en
modelos de ratones.
Soni menciona que la GDF-11 es una citoquina que bloquea la diferenciación terminal de los glóbulos rojos
mediante una amplificación paracrina que produce precipitación de las cadenas alfa. Por ende, empeora la
eritropoyesis ineficaz.
Desde hace varios años se estudia la utilidad terapéutica de fármacos que compiten con el ligado que se
une al dominio activo de los receptores asociados a estas moléculas de señalización. El objetivo es básica-
mente mejorar la maduración eritroide. En particular, la literatura menciona el luspatercept y sotatercept.
El estudio más reciente es uno de fase tres, doble ciego, controlado y randomizado, que exploró el porcen-
taje de reducción en la necesidad de transfusiones al usar luspatercept a una dosis de 1 - 1,25 mg/kg sub
cutáneos cada 21 días versus un grupo placebo. Mostró que 21.4% de los pacientes en el grupo de luspater-
cept alcanzó la meta de reducción mayor o igual al 33% de la necesidad de transfusiones de glóbulos rojos
comparado al placebo (4.5% de los pacientes). Episodios de dolor óseo, artralgias, hipertensión e hiperuri-
cemia fueron las razones reportadas en el estudio como justificantes de la interrupción del tratamiento (9).
En otro estudio fase dos, el beneficio de ese fármaco se evidenció también en un grupo de pacientes con
talasemia no dependiente de transfusiones, con mejoría en la hemoglobina (6).
El sotatercept también ha mostrado resultados beneficiosos tanto en pacientes dependientes de trans-
fusiones como no dependientes, aunque en estudios fase dos Taher y otros reportaron los resultados de
un estudio fase dos en el 2018 que examinó la efectividad del Ruxolitinib -un conocido inhibidor de la vía
JAK2-, con el fin de bloquear el eje Eritropoyetina - EPOR - JAK2 - STAT, limitar la proliferación excesiva de
progenitores eritroides en bazo, y así disminuir la esplenomegalia propia de la hematopoyesis extramedular
en estos pacientes.
Se evidenció una reducción sostenida en el tamaño esplénico en el grupo de pacientes que recibieron el
fármaco, que eran dependientes de transfusiones. El propósito principal del medicamento clínicamente es
53
mejorar la hemoglobina pretransfusional. Su uso también se reporta como seguro, aunque no hay estudios
fase tres (3).
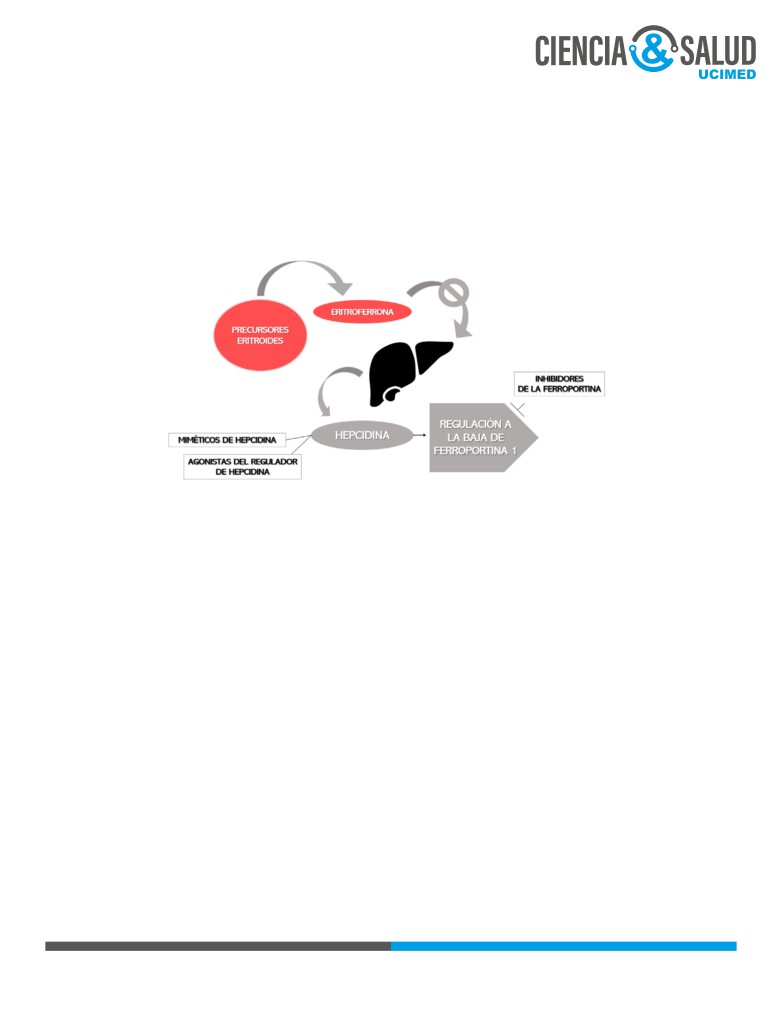
6.3 AGENTES MODULADORES DEL METABOLISMO DEL HIERRO
Estos fármacos se basan en el mecanismo de homeostasis de hierro asociado a la hepcidina. La hepcidina
es un péptido de síntesis hepática y reactante de fase aguda que regula el balance de hierro al influir en la
absorción y exportación de hierro celular (5).
FIGURA 2. Blancos farmacológicos asociados a la hepcidina. Elaboración por José Manuel Monge Ortiz, autor del artículo, 2020.
Como se observa en la figura 2, en las beta talasemias ocurre una supresión de la síntesis de hepcidina a
nivel hepático por el aumento de eritroferrona eritroide. Esto produce un aumento de la absorción de hierro
y por ende riesgo de sobrecarga de hierro sistémica.
Los fármacos en esta clase se encuentran aún en estudios fase I y II y corresponden a: miméticos de la hep-
cidina, agonistas del receptor Tmprss6 (regulador de hepcidina), e inhibidores de la ferroportina (6).
Los miméticos también son descritos en la literatura como mini hepcidinas. Son péptidos que inducen
efectos similares a la hepcidina de forma que disminuyen el hierro sérico y la sobrecarga férrica sistémica.
De momento, la hepcidina humana sintética LJPC-401 se evaluó en un estudio fase I mostrando efectos
prometedores (14).
En estudios pre clínicos, el inhibidor oral de la ferroportina VIT-2763 demostró adecuada biodisponibilidad
y capacidad in vitro para inhibir el movimiento de hierro celular de forma comparable con la hepcidina (15).
En el 2019, se inició un estudio fase I en sujetos sanos para evaluar la eficacia, farmacocinética, y farmaco-
dinamia del medicamento en dosis única y múltiples (14).
Asimismo, en junio del 2019, se inició un ensayo clínico que pretende estudiar la efectividad y seguridad
de la apo- transferrina humana en un total de doce pacientes con beta talasemia intermedia. Los detalles
pueden consultarse en la dirección electrónica clinicaltrials.gov.
Se postula que la apo transferrina puede disminuir los niveles lábiles de hierro sérico, normalizar la vida
media de los glóbulos rojos y mejorar la producción de hemoglobina en estos pacientes (14).
54
5. TERAPIA Y MANIPULACIÓN GÉNICA
La era de la secuenciación del genoma y la comprensión del cluster del gen de la beta globina, y su estricta
regulación, ha proporcionado nuevas opciones para el tratamiento de pacientes con talasemia. Actualmen-
te, se ha logrado comprender el switch en la producción de gamma a beta globina, así como los factores de
transcripción involucrados en este proceso (12).
El objetivo de la terapia génica para el tratamiento de la beta talasemia es lograr la introducción estable
de genes de globina funcionales en las propias células madre hematopoyéticas del paciente (HSC por sus
siglas en inglés) para corregir la eritropoyesis ineficaz y la anemia hemolítica, obviando así la necesidad de
transfusiones. Este método puede ofrecer la cura a aquellos que no pueden someterse a un trasplante alo-
génico o que carecen de un donante compatible (16).
Actualmente, los enfoques de terapia génica se pueden dividir en dos grupos amplios: 1. Adición de genes,
y 2. Edición de genes.
5.1 ADICIÓN GÉNICA
Este proceso implica la inserción de un vector lentiviral/retroviral que contiene todo el aparato regulador y
los genes productores de cadenas beta o gamma, en células madre humanas autólogas de manera in vitro,
y posteriormente la infusión de estas células madre modificadas de vuelta al paciente (en conjunto con
mieloablación). A pesar de que es conceptualmente fácil, el campo ha avanzado tecnológicamente solo re-
cientemente, pues ahora se pueden producir a gran escala los vectores (empaquetados con todos los genes
de beta globina) y con suficiente potencia para modificar un gran número de células madre humanas, y de
esta manera lograr proporcionar una respuesta clínica significativa (13).
Para una corrección duradera, las inserciones se realizan en células madre y progenitoras (células CD34 po-
sitivas). Los genes productores de globina deben colocarse bajo el control de un promotor eritroide especí-
fico, de modo que la transcripción de los genes insertados solo pueda darse en precursores eritroides y no
en otros tipos de células como glóbulos blancos o plaquetas. Una vez que una población suficientemente
grande de células madre ha sido modificada e infundida de nuevo al paciente, se espera que los progeni-
tores de eritrocitos derivados de estas células madre producirán suficientes cadenas beta (o gamma) para
combinarse con cadenas alfa y así reducir el desequilibrio que se presenta en estos pacientes (12).
Este enfoque tiene las ventajas obvias de la falta de complicaciones relacionadas con la histocompatibilidad
o la necesidad de agentes inmunosupresores. Además, el uso de un único producto aplicable a todos los
pacientes con beta talasemia independientemente de la mutación que presenten es un beneficio adicional.
(13).
Los primeros estudios en terapia génica se desarrollaron en los años noventa y principios de los años 2000
en modelos murinos. Sin embargo, no fue hasta el 2006 en Francia que se inició la primera prueba en huma-
nos, por el grupo Cavazzana-Calvo y Leboulch (patrocinado por la compañía Genetix Pharmaceuticals®).
El ensayo MSKCC fue el primer ensayo de terapia génica aprobado en los Estados Unidos en 2010, y todos
los pacientes demostraron marcadores génicos duraderos y estables. Posteriormente la compañía Blue Bird
Bio ® (BBB) financió cuatro estudios con el mismo vector, BB305. Estos son el ensayo NorthStar-HGB 204,
el HGB 205 y los ensayos HGB 207 y HGB 212, los cuales se encuentran en fase III. La terapia génica para
la beta-talasemia dependiente de la transfusión (TIGET-BTHAL) es un ensayo patrocinado por el Instituto
Teletón de Genética y Medicina, el cual finalizó en 2019 en Milán. El procedimiento fue bien tolerado y los
resultados hasta el momento indican una disminución en las transfusiones y una mejoría en la calidad de
vida (17).
55
5.2 EDICIÓN DE GENES
Nuevas tecnologías genéticas están surgiendo en el laboratorio. Por lo tanto, un conjunto de herramientas
de edición del genoma está creando nuevas perspectivas para impactar directamente mutaciones causan-
tes de enfermedad y secuencias reguladoras. Los últimos años han visto avances en la disponibilidad de
diferentes nucleasas - Nucleasas de dedo de zinc, Nucleasas efectoras tipo activador de la transcripción
y Repeticiones Palindrómicas Cortas Agrupadas con Intercalado Regular y Nucleasas asociadas a Crispr 9
(Crispr-Cas9 por sus siglas en inglés); todas estas son enzimas nucleasas que pueden cortar el ADN humano
en ubicaciones precisas (17).
Como fue descrito anteriormente, pacientes con talasemias que se presentan con niveles elevados de HbF
tienen un curso clínico mucho más favorable. La magnitud de la elevación de HbF afecta los síntomas de
las hemoglobinopatías en grados variables, pero incluso aumentos moderados pueden ser beneficiosos.
Parece ser que niveles de HbF superiores al 20% son suficientes para evitar crisis clínicas (17).
Es por ello que se pueden utilizar estas nuevas técnicas de edición genética para aumentar los niveles de
HbF. BCL11a (uno de los factores de transcripción que controla el cambio de HbF a HbA) es un objetivo
excelente para los enfoques de edición de genes. Al suprimir BCL11a, se postula que la producción de HbF
puede ser desencadenada nuevamente en pacientes con talasemia. Hacer deleciones específicas en el po-
tenciador eritroide de BCL11a es un enfoque prometedor que se está explorando actualmente. Otro método
para aumentar la producción de HbF mediante la edición de genes es recrear las mutaciones vistas en pa-
cientes con persistencia hereditaria de hemoglobina fetal (12).
Realizar estas pequeñas ediciones en las células madre de pacientes con talasemia de manera ex-vivo con
la herramienta Crispr-Cas9 puede proporcionar un tratamiento más económico en el futuro para mejorar
la enfermedad. A pesar de que este método parece prometedor, la introducción de terapias de edición del
genoma en la clínica requiere el abordaje de ciertos obstáculos, como la introducción de los agentes de
edición dentro de las células hematopoyéticas y la posible genotoxicidad (actividad enzimática fuera del
objetivo) (17)(18).
CONCLUSIONES
La beta talasemia es una condición relevante para el conocimiento médico por las extensas complicaciones
hematológicas, metabólicas y psicológicas que tienen en los pacientes. El impacto también se extiende al
sistema de salud, el cual debe ser capaz de proveer los suministros e intervenciones médicas oportuna-
mente, por lo que la investigación en nuevas terapias permite hacer un replanteamiento de la forma en que
tradicionalmente se han manejado estos pacientes.
El implemento de terapias menos cruentas con menos efectos adversos y con mayor especificidad augura
un mejor futuro para los pacientes con esta enfermedad, dados los muchos efectos adversos y complicacio-
nes del tratamiento actual. Las limitaciones en este campo recaen sobre el pobre conocimiento de algunos
mecanismos moleculares, así como de la ineficacia de estos tratamientos in vivo. Por el momento, aquellas
terapias centradas en reducir los efectos adversos por sobrecarga de hierro son estándar. Por ahora, las
terapias cuyo objetivo es aumentar la producción de hemoglobina fetal mediante fármacos o modificación
genética muestran resultados promisorios. Sin embargo, es necesaria la investigación continua y la actua-
lización de conocimientos de manera periódica para tener un panorama claro a la hora de abordar un pa-
ciente con esta patología.
56
BIBLIOGRAFÍA
1.
Origa R. β-Thalassemia.Genetics in Medicine [Internet]. 2016 [Citado 27/06/2020];19(6):609-619.
2.
Shah F, Sayani F, Trompeter S, Drasar E, Piga A. Challenges of blood transfusions in β-thalassemia,
d68ed.
3.
Cappellini M, Porter J, Viprakasit V, Taher A. A paradigm shift on beta-thalassaemia treat-
ment: How will we manage this old disease with new therapies? Blood Reviews [Internet]. 2018 [Citado
4.
Taher A, Karakas Z, Cassinerio E, Siritanaratkul N, Kattamis A, Maggio A et al. Efficacy and safety of
ruxolitinib in regularly transfused patients with thalassemia: results from a phase 2a study. Blood [Internet].
2018 [Citado 29/06/2020];131(2):263-265.
5.
Hoffman R, Benz E, Silberstein L, Heslop H, Weitz J, Anastasi J et al. Hematology. Philadelphia, PA:
Elsevier; 2018.
6.
Khandros E, Kwiatkowski J. Beta Thalassemia. Hematology/Oncology Clinics of North America [In-
ternet] 2019 [Citado 28/06/2020];33(3):339-353.
7.
de Dreuzy E, Bhukhai K, Leboulch P, Payen E. Current and future alternative therapies for beta-tha-
lassemia major. Biomedical Journal [Internet]. 2016 [Citado 25/06/2020];39(1):24-38.
8.
Salah, Naouel Ben, et al. Revisiting Beta Thalassemia Intermedia: Past, Present, and Future Pros-
pects. Hematology [Internet]. 2017 [Citado 1/08/2020];22(20):607-616.
9.
Hoffbrand A, Steensma, P. Essential Hematology. Philadelpia, PA: Wiley Blackwell, 2019.
10.
Cappellini M, Viprakasit V, Taher A, Georgiev P, Kuo K, Coates T et al. A Phase 3 Trial of Luspatercept
in Patients with Transfusion-Dependent β-Thalassemia. New England Journal of Medicine. [Internet] 2020
11.
Betts, Marissa, et al. Systematic Literature Review of the Burden of Disease and Treatment for Trans-
fusion-Dependent β-Thalassemia. Clinical Therapeutics [Internet]. 2020 [Citado 29/06/2020]; 42(2):322-
337.
12.
Soni S. Novel and innovative approaches for treatment of β-thalassemia. Pediatric Hematology On-
cology Journal [Internet]. 2017 [Citado 29/06/2020];2(4):121-126.
13.
El-Beshlawy, Amal, and Mona El-Ghamrawy. Recent Trends in Treatment of Thalassemia. Blood
Cells, Molecules, and Diseases [Internet].
bcmd.2019.01.006
57
14.
Thein S. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells, Molecules, and
15.
Richard F, Lier J, Roubert B, Haboubi T, Göhring U, Dürrenberger F. Oral ferroportin inhibitor
VIT-2763: First-in-human, phase 1 study in healthy volunteers. American Journal of Hematology [Internet].
2019 [Citado 29/06/2020];95(1):68-77.
16.
Karponi G, Zogas N. Gene Therapy for Beta-Thalassemia: Updated Perspectives. The Application
S178546
17.
Boulad, Farid, et al. Gene Therapy and Genome Editing.” Hematology/Oncology Clinics of North
18.
Wienert, Beeke, et al. Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopa-
tig.2018.09.004
58