Enfermedad atípica: Inmunodeficiencia combinada grave.
Atypical disease: Severe Combined Immunodeficiency.
Mildred Angélica Sauce Guevara 1, Erika Palacios Rosas2.
1 y 2 Universidad de las Américas Puebla, México
Contactos: erika.palacios@udlap.mx mildred.saucega@udlap.mx
RESUMEN
La inmunodeficiencia combinada grave (IDCG) es una enfermedad hereditaria ligada
al cromosoma X en donde existen anomalías en las funciones de los linfocitos T, B y
NK, que puede ser mixta y afectar a dos o más de estas poblaciones celulares. Debido
a que la inmunidad adaptativa está abolida, los pacientes con esta afección son
susceptibles a infecciones recurrentes por patógenos oportunistas y no oportunistas,
que resulta en la muerte temprana a menos que la reconstitución inmunitaria sea
posible. Aún no existe un registro amplio de esta enfermedad dado que es de muy
baja incidencia y a pesar de no tener cura, su pronóstico es relativamente favorable
si es detectada a tiempo. Actualmente, existen algunos tratamientos para mejorar la
calidad de vida del paciente como el trasplante de la medula ósea, la terapia génica
o el remplazo de la inmunoglobulina intravenosa (IGIV).
El objetivo principal de esta revisión consiste en exponer aspectos adicionales de
manera clara y concisa sobre cuestiones básicas para la mejor comprensión de esta
enfermedad.
Palabras Clave: anomalías, congénitas, inmunodeficiencia.
ABSTRACT
Severe combined immunodeficiency (SCID) is an inherited disease linked to the X
chromosome where there are abnormalities in the functions of T, B and NK lymphocytes,
Cómo citar:
which can be mixed and affect two or more of these cell populations. Because adaptive
Sauce Guevara , M. A.,
& Palacios Rosas, E.
immunity is abolished, patients with this condition are susceptible to recurrent infections
Enfermedad atípica:
by opportunistic and non-opportunistic pathogens, resulting in early death unless
Inmunodeficiencia
immune reconstitution is possible. There is still no extensive registry of this disease,
combinada grave.
Revista Ciencia Y Salud,
given that it has a very low incidence and despite having no cure, its prognosis is
doi.org/10.34192/
relatively favorable if it is detected early. Currently there are some treatments to
cienciaysalud.v6i1.392
improve the quality of life of the patient such as bone marrow transplantation, gene
therapy or the replacement of intravenous immunoglobulin (IVIG).
The main objective of this review is to present additional aspects in a clear and concise
manner on basic questions for a better understanding of this disease.
Keywords: Pneumonia; abnormalities, congenital, immunodeficiency.
21/oct/2021
14/ene/2022
14/Feb/2022
6
63
Conflicto de intereses
Los autores manifiestan que no existe ningún conflicto de intereses.
Financiamiento
No existen fuentes de financiación públicas o privadas en la realización del presente estudio.
INTRODUCCIÓN
La inmunodeficiencia combinada grave (IDCG) es un conjunto de enfermedades del sistema inmunitario que
tienen las características de ser hereditarias, además de presentar anomalías en el funcionamiento de los
linfocitos T, B y NK, aunque en algunos casos esta puede ser mixta por lo que puede afectar de dos a más
de dichas poblaciones celulares.1 La IDCG pertenece al grupo llamado: Inmunodeficiencias Primarias (IDP),
que reportan más de 200 diagnósticos diferentes y la mayor parte son trastornos derivados de anomalías
o defectos monogénicos que afectan el desarrollo del sistema inmune.
La IDCG se considera como una enfermedad extremadamente rara o poco común debido a que su frecuencia
varía de 1/50 000 a 1/1 000 000 de recién nacidos vivos2 lo que ocasiona que sea frecuentemente
subdiagnosticada o subregistrada.3 Es necesario por tanto que se siga documentando información dirigida
al personal de salud para su actualización sobre este tipo de patología en especial.
Figura 1: Diagrama paciente sano vs paciente con IDCG.(Elaboración propia)
METODOLOGÍA
La búsqueda bibliográfica se realizó a través de la consulta en: GOOGLE ACADÉMICO, MEDLINE PLUS y
PUBMED con los siguientes criterios de selección: severe combined immunodeficiency AND pathophysiology
AND disease y delimitando los tiempos de publicación entre los años 2001 al 2021. Para la realización del
artículo se seleccionaron 22 resultados que hacían referencia a la explicación de la enfermedad, se llevó
acabo la lectura de cada uno de ellos para la discriminación y la integración de la información descrita.
64
DESARROLLO
EPIDEMIOLOGÍA
No existe algún registro central que contenga el número de pacientes diagnosticados con IDCG, pero las
estimaciones encontradas oscilan entre 40 y 100 pacientes, por lo que la IDCG se considera una enfermedad
extremadamente rara, con variación regional además de presentar mayor incidencia entre poblaciones
con altas tasas de consanguinidad.4 Por otro lado, no existen registros claros sobre el número de niños no
diagnosticados que mueren cada año en el mundo por infecciones relacionadas con esta patología, pero se
estima que puedan existir algunos decesos por IDCG no relacionadas por dichas infecciones,3 por otra parte,
un estudio realizado por Chan, K., et. al., demostró que el diagnóstico tardío del IDCG provocó el deceso de
20 infantes de una población de 39 pacientes diagnosticados.5 Es por esta razón que se han implementado
sistemas de tamizaje neonatal en Estados Unidos que han permitido exhibir de manera objetiva la incidencia
de la enfermedad, aunque es necesario que en otros países sea integrado este programa de tamizaje neonatal
en los sistemas de salud.6
FISIOPATOLOGÍA
El fenotipo de los linfocitos funcionales depende de una mutación específica y está determinado por la
fisiopatología, el estado inmunológico y la susceptibilidad a tipos específicos de infecciones en pacientes
con la IDCG. Varias clasificaciones de la IDCG están sujetas a varios factores y la subclasificación tradicional
de la IDCG en: T-B+NK-, T-B-NK+, T-B+NK+, o T-B-NK-, se formó después de la identificación de las líneas
de linfocitos afectados. 7
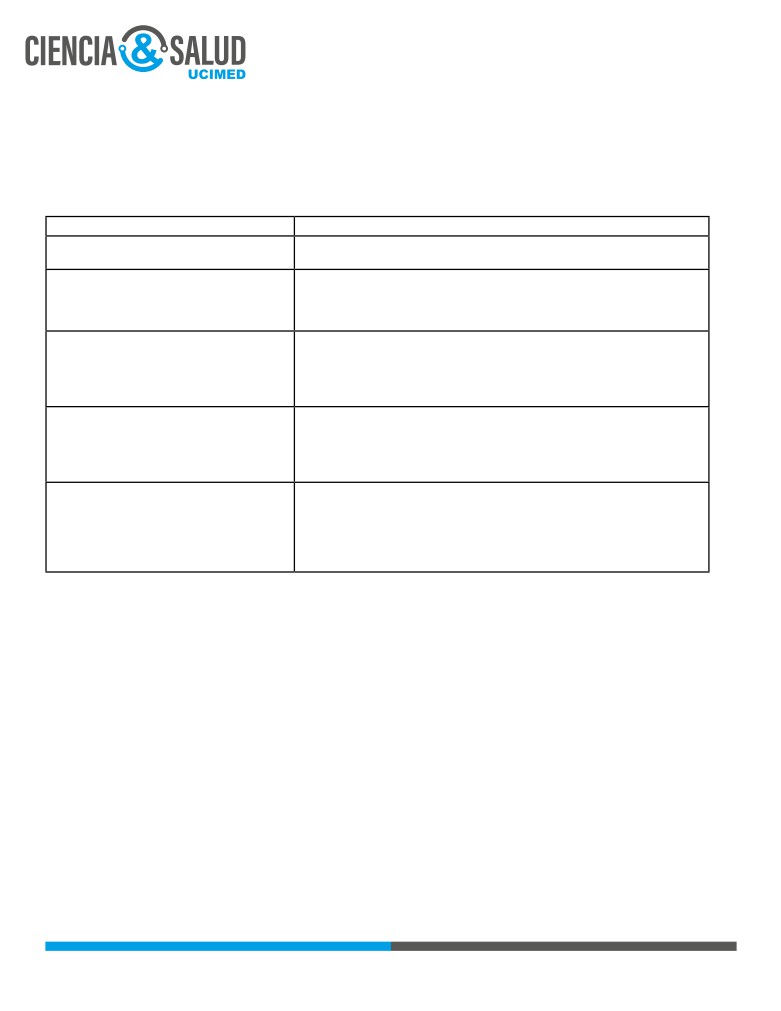
En la tabla 1 se muestra la subclasificación tradicional del IDCG:
T-B+NK-
Su aparición está influenciada por defectos moleculares que afectan a los linfocitos
T y linfocitos NK. El gen más afectado es IL2RG, puntualmente en el cromosoma
Xq13 que codifica la cascada gamma y sus receptores comunes que son: IL2, IL-4,
IL-7, IL-9, IL-15 e IL-21. Por ejemplo, la activación del receptor IL-7 es necesario para
la maduración de las células T en el timo; mientras que la activación del receptor
de IL-15 estimula la supervivencia de las células NK.8
T-B-NK+
La recombinación de receptores es un proceso al que deben someterse los linfocitos
B y los linfocitos T durante su desarrollo. Cuando este proceso falla, el resultado es
la apoptosis. Las proteínas 3RAG1 y 3RAG2 cortan sitios de ADN que permiten la
recombinación. Después del corte, se forma un bucle entre las cadenas del ADN,
mientras que la ARTEMIS dirige el bucle de ADN para recombinar sus fragmentos.
Cualquier disfunción de estos mecanismos es la causa de este fenotipo. 9
T-B+NK+
El mecanismo de prevención de la apoptosis de células T es la activación de las vías
del receptor de TCR e IL-7. El receptor de IL-7 consta de varias secuencias como
son: las cadenas gamma y las cadenas alfa. Cuando la cadena alfa se ve afectada
selectivamente, las células T alteran su viabilidad. Por otro lado, el CD3 es una
molécula complementaria a la señalización del TCR, por lo que cuando una de las
hebras que la componen tiene una mutación, esta no sobrevivirá. 10
T-B-NK-
Este es considerado el fenotipo más grave, debido a que la función de los linfocitos
se vuelve deficiente en todas las generaciones descendientes de la persona que
lo presenta. Los linfocitos son células que contienen una alta tasa de metabolismo
y reproducción por lo que cuando se altera la función de la ADA (adenosina
desaminasa) esencial para la vía de recuperación de las purinas, se produce la
acumulación de metabolitos tóxicos lo que conduce a la toxicidad y posteriormente
a la muerte celular. 10
65
ETIOLOGÍA
La causa de esta enfermedad está asociada con el cromosoma X que contiene una mutación en el gen IL2RG
(Xq13) y que a su vez codifica la secuencia gamma común. Hasta la fecha, se han identificado mutaciones
en 15 genes IDCT que son genéticamente recesivos11.
La Tabla 2 muestra las mutaciones presentes en la IDCG:
Mutación
Explicación
T-B-NK-IDCG
Este tipo de mutación causa una diagénesis reticular debido a que existe
una falta inminente de células madre.
T-B-NK+IDCG (defecto RAG1/2)
Este tipo de mutación puede inducir la IDCG ya que las enzimas RAG1/ 2
escinden el ADN para reorganizar el VDJ del TCR y del BCR. Un fenotipo
similar es la deficiencia de Artemis, que se caracteriza por la falla de la
reparación del ADN después de la escisión de RAG1 / 2.
T-B+NK-IDCG
La IDCG puede estar ligada al cromosoma X y en ausencia del receptor
de IL para muchos tipos de células diferentes debido a la falta de una
cascada gamma común. Un fenotipo similar conocido como deficiencia
de quinasa Jak 3 ocurre debido a una deficiencia de quinasas de
señalización Jak 3 a través de la unión de IL-R.
T-B+NK+IDCG
Este tipo de mutación presenta un fenotipo de deficiencia de IL-7 en
ausencia de la cadena alfa de IL-7 lo que conduce al fracaso de la
diferenciación de las células T. Existe un fenotipo similar en presencia de
IL-7 en donde aparece una inactivación de CD3 y se caracteriza por una
transducción de señal defectuosa, por ejemplo: la deficiencia de ZAP-70.
MHCT+B+NK+
Esta mutación conduce a una falla en el fenotipo mediante dos
condiciones: deficiencia de MHC de clase I (síndrome de linfocitos
desnudos) y deficiencia de MHC de clase II, en donde la primera
condición se debe a la falta de expresión del MHC de clase I debido a
un defecto en la transcripción de TAP-2 y en el último caso es causado
por un defecto transcripcional de la proteína MHC II. 12
MANIFESTACIONES CLÍNICAS
Entre las manifestaciones clínicas más recurrentes se presentan infecciones graves frecuentes y/o
potencialmente fatales, retrasos en el crecimiento, diarrea crónica, infecciones respiratorias regulares y
usualmente los pacientes en sus primeros meses de vida sufren de serios peligros que pueden afectar el
desarrollo de una buena calidad de vida.
Además de las anteriores, se pueden manifestar verrugas cutáneas persistentes, celulitis, eccema atípico,
lesiones de musgo contagioso, pérdida de cabello y dermatitis seborreica grave, además, se puede presentar
hepatitis crónica o colangitis esclerosante. En estos pacientes, las infecciones bacterianas oportunistas, como los
hongos de Pneumocystis Jiroveci, Candidiasis y Cryptosporidium, son comunes. También se pueden encontrar
microorganismos intracelulares, como la bacteria de listeria, Salmonella Typhi, toxoplasma y mycobacterium,
infección de aspergillus o virus como adenovirus, virus sincitial respiratorio (VSR), citomegalovirus (CMV),
virus del herpes (HSV) o Epstein Barr (EBV). La sospecha de la IDCG todavía se considera una “emergencia
pediátrica” debido a que se presenta en los primeros meses de vida y corre el riesgo de desarrollarse
rápidamente desencadenando el deceso del paciente13.
DIAGNÓSTICO
El diagnóstico principal de esta enfermedad se confirma mediante la historia clínica. En ella se deben
considerar las afecciones que haya sufrido el paciente tales como: ocho o más infecciones de oído, dos o
66
más casos de neumonía, infecciones que no resueltas con el tratamiento durante un período de dos meses
o más, que haya experimentado poco o ningún aumento de peso y estatura, infecciones que requieren
tratamiento con antibióticos intravenosos, infecciones más profundas como la neumonía que afectan a toda
el área del pulmón o absceso hepático, aftas persistentes en la boca o garganta, antecedentes familiares de
inmunodeficiencia o presencia de muerte infantil por infección en familiares cercanos.
Si un paciente tiene 3 o más de estas afecciones, el diagnóstico se confirma mediante análisis de sangre a
través de recuentos de células T y B, que muestran niveles relativamente bajos de células T y anticuerpos,
como la inmunoglobulina14.
En pacientes con inmunodeficiencia combinada grave, la concentración total de linfocitos suele estar por
debajo de lo normal, oscilando desde un límite indetectable de hasta 0,005×106 células/mm3. Sin embargo,
el hecho de que estos valores bajos no estén presentes no descarta el diagnóstico positivo a esta patología
debido a que algunos defectos genéticos pueden afectar la función de los linfocitos sin afectar su número.
La prueba más precisa para determinar la función de las células T implica colocar las células sanguíneas del
infante en tubos de cultivo, tratarlas con varios estimulantes e incubarlas durante varios días. Las células T
normales responden a los estímulos desencadenando la división celular. Por el contrario, los linfocitos de los
pacientes con la IDCG generalmente no responden a estos estímulos15-16.
Otro diagnóstico de esta patología se puede obtener antes del nacimiento. Esto se hace en los casos en
el que hay un primer hijo en la familia con esta afección y si se ha identificado un defecto genético. Si se
han realizado pruebas genéticas de infantes previamente afectados, el diagnóstico se puede confirmar en
embarazos posteriores mediante pruebas moleculares. Las tres principales estrategias de secuenciación de
próxima generación son:
La secuenciación de panel genómico, en la que se secuencia una lista específica de genes previamente
identificados (todos los genes IDCG conocidos). Es una opción atractiva cuando los fenotipos clínicos e
inmunológicos sugieren claramente esta enfermedad.
La secuenciación del exoma completo, donde el fenotipo clínico es atípico y todas las regiones del ADN son
codificantes para la formación proteica (exones) y en donde se concentra aproximadamente el 85% de las
variantes genéticas que pueden causar alguna patología conocida actualmente.
La secuenciación del genoma completo, incluido el análisis de exones e intrones. Además de estos métodos
de diagnóstico, las células se pueden identificar a partir de una muestra de vellosidades coriónicas (CVS) en
la placenta o mediante amniocentesis de la que se extrae una pequeña cantidad de líquido amniótico de la
cavidad uterina para determinar la IG17-18.
TRATAMIENTOS DISPONIBLES
El tratamiento inicial de los pacientes con la IDCG se determina con base en el fenotipo, obtenido a través de la
sospecha clínica e informes de laboratorio que evalúan el sistema inmunológico cuantitativa y cualitativamente,
no obstante, en México, América Latina y muchas partes del mundo todavía existen límites para un diagnóstico
genético rápido, una condición que puede retrasar el tratamiento y la toma de decisiones. 19
Trasplante de células progenitoras hematopoyéticas
El propósito del trasplante es reemplazar las células disfuncionales del paciente (receptor) con células
funcionales obtenidas de un donante sano compatible con antígenos leucocitarios humanos (HLA).
El éxito del trasplante depende de varios factores, ahora identificados, que mejoran el pronóstico de los
pacientes con inmunodeficiencia combinada grave como son: el diagnóstico oportuno, la ausencia de infección
activa en el momento de la intervención, el tratamiento completo antes, durante y después del trasplante,
67
el mayor grado de histocompatibilidad de los tejidos del donante con el receptor y tener la disponibilidad
de donantes de células madre hematopoyéticas. 20
TERAPIA GÉNICA
La aplicación de ácidos nucleicos se lleva a cabo con fines terapéuticos, actualmente existen muchas
estrategias en la investigación clínica y en el desarrollo de esta tecnología por lo que se ha descrito que
este tipo de tratamiento ha demostrado ser clínicamente eficaz especialmente para los defectos de IL2RG
y ADA provocados por la IDCG. 21
Reemplazo de inmunoglobulina intravenosa
En este tipo de tratamiento se lleva a cabo la reposición de los anticuerpos que fueron afectados por los
defectos de las células B, pero siempre se debe considerar que la función de las células T defectuosas no
se podrán restaurar.22
PRONÓSTICO
La IDCG debe ser manejada por un equipo de expertos como pediatra, genetista, inmunólogo clínico,
enfermera, farmacéutico y un especialista en enfermedades infecciosas para prevenir futuras complicaciones
y asegurar que la calidad de vida del paciente sea adecuada. El equipo de tratamiento también puede brindar
asesoramiento a los padres, para los cuidados del paciente con IDCG.23 La IDCG tiende a tener el peor
pronóstico a menos que se realice con éxito algún tipo de tratamiento ya que si la IDCG es diagnosticada
antes de los 3 meses de edad, la supervivencia posterior al trasplante de células madre hematopoyéticas
de cualquier tipo es del 95%.24-25.
En general, para mejorar la calidad de vida de los pacientes con IDCG, se necesita la administración a largo
plazo de medicamentos en su mayoría antimicrobianos además de requerir de un manejo de historial clínico
del paciente personalizado.26
CONCLUSIONES
La IDCG es un síndrome que trae consigo diversas manifestaciones clínicas variables, que pueden llegar a
dificultar su diagnóstico y por lo tanto no permite brindar un tratamiento oportuno al paciente traduciéndose
en una disminución de la calidad de vida. Además, la importancia de identificar y conocer de una manera
general las implicaciones de la IDCG es necesaria dado que es una de las enfermedades poco comunes
y subregistradas en México y en el mundo por lo que es conveniente tener información adicional para
incrementar y recordar el conocimiento que se tiene al respecto.
REFERENCIAS BIBLIOGRÁFICAS
1 Justiz Vaillant AA, Mohseni M. Severe combined immunodeficiency. En: StatPearls. Treasure Island
(FL): StatPearls Publishing; 2020.
2 Murguía Pérez JG, Pérez-Gaxiola G, García-Domínguez M. Inmunodeficiencia combinada grave:
informe de caso. Alergia, Asma e Inmunología Pediátricas. 2020;29(1):37-41.
3 Saucedo Aparicio AG, Espinosa PSE, González SME, et al. Inmunodeficiencias combinadas graves,
¿enfermedades raras o subregistradas? Alerg Asma Inmunol Pediatr. 2018;27(2):37-43
4 Orphanet, D. L. T. U. I. Orphanet: Inmunodeficiencia combinada grave. [Internet]. [citado el 7 de febrero
68
5 Chan K, Davis J, Pai SY, Bonilla FA, Puck JM, Apkon M. A Markov model to analyze cost-effectiveness
of screening for severe combined immunodeficiency (SCID). Molecular Genetics and Metabolism.
2011;104(3), 383-389.doi:10.1016/j.ymgme.2011.07.007.
6 Vogel, B. H., Bonagura, V., Weinberg, G. A., Ballow, M., Isabelle, J., DiAntonio, L.,Caggana, M. Newborn
Screening for SCID in New York State: Experience from the First Two Years. Journal of Clinical
Immunology. 2014;34(3), 289-303. doi:10.1007/s10875-014-0006.
7 Acerca de la inmunodeficiencia combinada grave [Internet]. Genome.gov. [citado el 20 de octubre de
8 Castro SR, Padilla VER. Tamizaje de inmunodeficiencia combinada grave y su oportunidad para
implementarse en México [Internet]. Medigraphic.com. [citado el 9 de agosto de 2021]. Disponible en:
9 Kohn LA, Seet CS, Scholes J, Codrea F, Chan R, Zaidi-Merchant S, et al. Human lymphoid development
in the absence of common γ-chain receptor signaling. J Immunol. 2014;192(11):5050-8.
10 Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, et al. A founder mutation in Artemis, an SNM1-
like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002;168(12):6323-9.
11 Sewell WAC, Khan S, Doré PC. Early indicators of immunodeficiency in adults and children: protocols
for screening for primary immunological defects. Clin Exp Immunol. 2006;145(2):2013.
12 Superusuario. Pruebas genéticas - Inmunodeficiencia combinada severa ligada a X (inmunodeficiencia
combinada severa ligada al cromosoma X) - Gen IL2RG. - IVAMI [Internet]. Ivami.com. [citado el 10
de-geneshumanos-enfermedades-neoplasias-y-farmacogenetica/1689-pruebas-geneticas-
inmunodeficiencia-combinada-severa-ligada-axx -inmunodeficiencia-combinada-grave-vinculada-
gen-i-il2rg-i.
13 Justiz Vaillant AA, Mohseni M. Inmunodeficiencia combinada grave. En: StatPearls. Treasure Island
(FL): StatPearls Publishing; 2020.
14 Martín del Campo Rodríguez LE, Sifuentes Osornio J. Infecciones oportunistas en el síndrome de
inmunodeficiencia adquirida: La historia en México a 20 años del inicio de la epidemia. Rev Invest Clin.
2004; 56 (2): 169-80.
15 Inmunodeficiencia combinada grave [Internet]. Nih.gov. [citado el 10 de agosto de 2021]. Disponible
16 Fischer A, et al. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers
17 Coria-Ramírez E, et al. Panorama epidemiológico de las inmunodeficiencias primarias en México.
Rev Alerg Mex 2010;57(5):159-63.
69
uploads/2017/07/INMUNODEFICIENCIA-COMBINADA-GRAVE_06.02.08.
19 Severe Combined Immunodeficiency and combined immunodeficiency [Internet]. Primaryimmune.
disease/severe-combined-immunodeficiency-and-combined-immunodeficiency.
20 Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP).
21 Cavazzana M, Touzot F, Moshous D, Neven B, Blanche S, Fischer A, “Stem cell transplantation
for primary immuno- deficiencies: the European experience.” Curr Opin Allergy Clin Immunol. 14
Dec;14(6):516-20.
22 Titman P, et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell
transplantation for severe congenital immunodeficiencies. Blood 2008;112(9):3907-3913. http://doi.
org/10.1182/ blood-2008-04-151332.
23 Justiz VA, Mohseni M. NCBI - Severe Combined Immunodeficiency. NCBI. [Internet]. [citado el 6 de
s11_.
24 Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the
diagnosis and management of primary immunodeficiency. Annals of allergy, asthma & immunology:
official publication of the American College of Allergy, Asthma, & Immunology. 2005 May;94(5 Suppl
1):S1- 63. PubMed PMID: 15945566.
25 Lipstein EA, Vorono S, Browning MF, Green NS, Kemper AR, Knapp AA, et al. Systematic evidence
review of newborn screening and treatment of severe combined immunodeficiency. Pediatrics.
2010;125(5):e1226-35.
26 Chinn IK, Shearer WT. Severe Combined Immunodeficiency Disorders. Immunology and Allergy
Clinics of North America. 2015; 35(4), 671-694.doi:10.1016/j.iac.2015.07.002.
70